
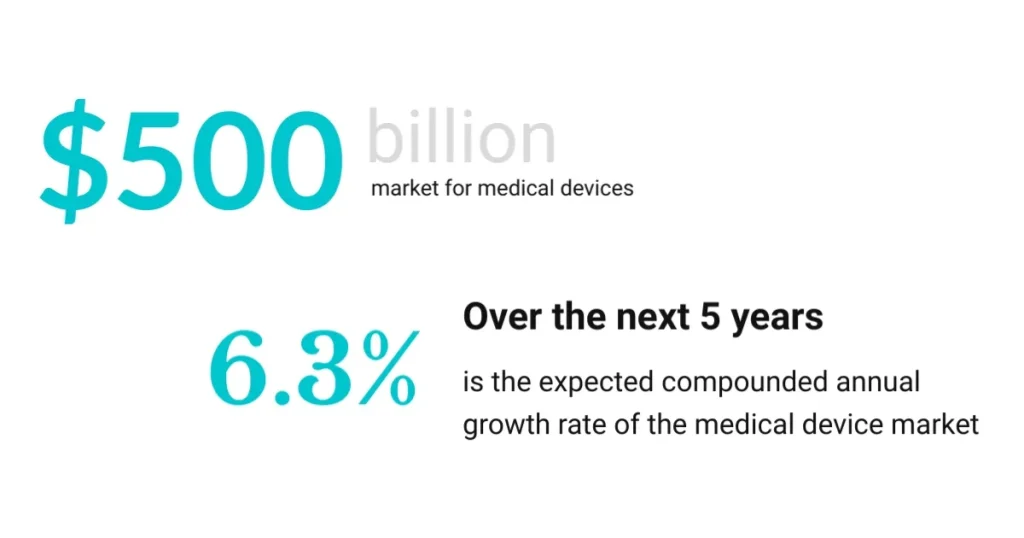
It’s no secret that understanding the Medical Device Development Timeline is crucial as the medical device industry is booming. Today, the global medical device market is valued at around $500 billion. Combined with a compound annual growth rate (CAGR) of 6.3% in the next five years, it’s an industry that shows no signs of slowing down. Driving this trend is a combination of sophisticated medical knowledge and cutting-edge tech.
But bringing new devices to market is tricky. Navigating the process requires a thorough understanding of the medical device development timeline under the United States Food and Drug Administration ( US FDA ).
In this article, we walk you through the medical devices research and development process, step-by-step. A solid grasp of these 8 fundamental steps will enable you to better communicate with your team, effectively meet FDA compliance regulations, and streamline your device’s time-to-market (TTM).
What Is a Medical Device?
A medical device is any product designed for diagnostic or therapeutic healthcare applications. This usually takes the form of an instrument or an implant, an apparatus or a machine. Some medical devices (like tongue depressors) are simple. Others (like implanted cerebral stimulators) are complex.
Healthcare professionals depend on an array of medical devices to monitor, diagnose, and treat patients. Whether a device is simple or highly complex, each must comply with strict regulations to ensure the utmost safety and efficacy. Likewise, each follows a similar product development process . Let’s now look at those 8 stages to develop medical device technology.
Medical Device Development Timeline in 8 Steps
Understanding and adhering to the eight major steps of development will have an enormous impact on the success of your medical device. Below, we break down exactly what you need to know about each stage in the process.
Step 1 – Discovery
Every medical device product starts as a good idea. That’s why the discovery phase is the first stage of the medical device development process . The goal is simple: Develop a viable concept.
Of course, generating a good idea requires an extensive investigation into unmet clinical needs, user requirements, risk analysis , market potential, and existing competition. This will help to determine whether a concept is technically sound and commercially feasible.
The discovery phase often involves evaluating multiple concepts and selecting the most promising one to move forward with. Once a concept is accepted, it’s important to map out its basic features, specs, and any potential regulatory requirements.
Learn more about the medical device development process on our blog:
- 10 Steps in Medical Device Design
- Navigating EU MDR
- The Difference Between Class I and Class II FDA Regulated Devices
Put simply, this stage begins with brainstorming and ends with a successful proof of concept and early design controls .
Step 2 – Design and Engineering
Once the concept has been defined, the design and development phase can begin. This involves creating detailed specifications for the device, followed by medical device prototyping for testing. Once a design is mature, a prototype goes into design freeze and is ready for medical device testing. These tests—known as verification and validation —ensure that the product functions as it should.
During this phase, it’s essential to consider things like quality, usability, safety, scalability, and regulatory requirements. With respect to the latter, be ready to prepare documentation that demonstrates how your product meets the regulatory requirements in order to receive approval from the FDA.
Step 3 – Preclinical Testing
Before a medical device can be studied in humans, it must undergo preclinical testing to ensure its safety and efficacy.
This phase of testing typically involves animal studies, although some devices may undergo simulated use testing, bench testing, or in-vitro testing. The objective is to identify any potential safety issues that may adversely affect human subjects.
Step 4 – Clinical Testing
If your medical device passes preclinical testing, it can move on to clinical testing. This phase involves testing the device on human subjects to determine how safe and effective it is.
Each clinical trial must conform to strict regulations, including obtaining informed consent from participants and reporting any adverse events to regulatory authorities.
Trials are usually conducted in three phases , aptly referred to as Phase I, Phase II, and Phase III. Together, these three phases offer mounting evidence to demonstrate your medical device’s safety, effectiveness, and usability in real-world patient populations.
Step 5 – Regulatory Approval
Once the clinical testing phase is complete, your team will need to prepare and submit a medical device application for regulatory approval. Documentation will consist of items such as clinical trial data, design information, manufacturing processes, and labeling.
Regulatory bodies vary by country. But, in the US, the FDA is responsible for regulating the medical device industry. Therefore, upon receiving FDA approval for your device, you will be legally allowed to begin commercialization.
Step 6 – Manufacturing and Quality Control
Now that your device has met FDA standards, it can enter the manufacturing and quality control phase. Broadly speaking, this involves setting up a medical device manufacturing process that adheres to FDA regulatory requirements and ensures the consistent quality of your product. For long-term viability, your process needs to be both safe and scalable.
A quality management system (QMS) must be put into place to ensure good manufacturing practices . This involves testing and inspecting the device at various stages throughout the manufacturing process, prior to its release for sale.
Remember, quality control is an ongoing process and any QMS should be ISO 13485 -compliant. This ISO standard provides a framework that ensures your device meets international compliance standards.
Step 7 – Post-Market Surveillance
Once a device is available to patients and healthcare providers, it’s essential to monitor its performance and address any issues that arise.
Have More Confidence In Your Medical Device Design
Our proven design process includes several iterations of prototyping to ensure your device functions as expected before moving into full scale production.
Post-market surveillance is therefore largely a process of risk management . It involves collecting data on the device’s safety and effectiveness, developing and implementing corrective and preventive actions (CAPAs) to address identified issues, and reporting any adverse events to regulatory authorities.
Step 8 – Continued Development
A lot of medical device development carries on through the entire product life cycle. Over time, manufacturers may continue to refine and improve their products. This is especially true for class II devices .
Continued development may involve developing new features or capabilities, improving the device’s performance or safety, or making other modifications based on feedback from users and healthcare providers.
Depending on the significance of the change or improvement, a letter to file with the FDA is needed or in extreme cases, a full resubmission is required.
As we’ve seen, the medical device product development life cycle is a lengthy and detailed process. But, by following the eight steps outlined above, medical device manufacturers can successfully bring medical device products to market.
Remove the Guesswork From Your Medical Device Development Timeline, With Vantage MedTech
Medical device development is a complex and highly regulated process that requires a big investment of time, resources, and expertise. In fact, from concept to market, the development timeline can span several years.
To ensure a smooth timeline, it’s important for manufacturers to plan and manage the process effectively. The goal must always be to ensure the device’s safety and efficacy. But that’s often easier said than done.
Looking to streamline your development timeline and eliminate errors? Partner with Vantage MedTech . For nearly three decades, we’ve helped companies like yours navigate the choppy waters of medical device development. Our services include:
- Medical device design and prototyping
- Medical device design engineering
- Medical device manufacturing
- Medical device regulatory compliance
- And every step in between.
Contact us today to streamline your medical device development timeline .

Time To Market
According to research, time-to-market is a primary competitive driver in the medical device industry, second only to innovation.* You can’t start making money until you get FDA approval for your medical device.
So, how long does it really take for the FDA to approve a medical device? The short answer: anywhere from one week to eight months, depending on the device class and a range of other factors.
Approval Process
To give you a better sense of how long it takes for the FDA to approve a medical device, let’s delve into the basics about the FDA’s medical device approval process as it pertains to each of the three classes:
- Class I medical devices have a low impact on patients’ overall health and are subject to the fewest regulatory requirements
- Class II devices are moderate-risk devices that require a Premarket Notification 510(k)
- Class III devices follow rigorous controls, most of which require a Premarket Approval (PMA)
Process by Class
The majority of Class I devices can be registered and listed with the FDA, which offers the fastest path to approval—as short as a week or two. Class II is a bit more complicated. First, you’ll need to submit a 510(k) application demonstrating that your device is Substantially Equivalent (SE) to one or more predicate devices previously cleared by the FDA. Once the submission is received, the CDRH or CBER’s Document Control Center (DCC) will assign the unique submission control number.
If the proper user fee has been paid and a valid eCopy has been provided, the DCC will then email an Acknowledgement Letter that notes the date they received the submission and the number assigned to it. The submission is then assigned to a Lead Reviewer, who will conduct the Acceptance Review, and if it meets the minimum threshold of acceptability, they will then respond according to the Refuse to Accept (RTA) Policy, within 15 days.
Possible Results
At the end of the 15 days, you will receive the Acceptance Review results. This will include the name and contact information of the Lead Reviewer assigned to the 510(k) and the status of the 510(k), which will be one of the following:
- The 510(k) was accepted for substantive review; or
- The 510(k) was not accepted for review (i.e., considered refused to accept or RTA); or
- The 510(k) is under substantive review because the FDA did not complete the acceptance review within 15 calendar days.
The status that’s assigned will directly impact how long it takes for the FDA to approve your medical device. If the FDA refuses the submission, the application will be put on RTA Hold for up to 180 days. At this point, you’ll have 180 days to fully address the deficiencies cited in the RTA Hold. If it is not done, the 510(k) is considered withdrawn and deleted from the system. If it is deleted, a new 510(k) must be submitted to obtain market clearance.
Substantial Review Process:
When considering how long it will take for the FDA to review your medical device, don’t forget to factor in the substantial review process, which begins as soon as the submission is accepted. At this time, the FDA may reach out to you with an Interactive Review to address “easy to fix” items or obtain information that may be adequately addressed within the timeframe set by the Medical Device User Fee Amendment (MDUFA) performance goal for 510(k) (90 FDA days). If the Lead Reviewer sends an AI Request, the submission is placed on hold, at which point you have 180 days to submit a complete response to the AI Request.
More about the Substantive Review Process:
To get a better idea of how long it will take the FDA to approve your medical device, keep the following in mind: First, any correspondence with the FDA during the Substantive Review process should be reviewed in a timely manner. Second, even if you do provide additional information as requested, don’t assume the FDA won’t issue a formal Additional Information (AI) request.
Be sure to submit all requested information as soon as possible; the FDA will not review it until it is received in its entirety, which will only add to the time it takes for the FDA to approve your medical device. After the AI Request is submitted, they will issue an SE or NSE letter for the submission. As noted earlier, the CDC must receive your response within 180 calendar days of the date of the AI Request or the submission will be withdrawn and deleted. No extensions beyond 180 days is granted. To minimize the time it takes for the FDA to approve your medical device, it is imperative that you avoid going beyond this critical 180-day mark.
510(k) Decision Letter:
The FDA will issue the decision letter by email to the address provided in the 510(k) cover letter. A 510(k) that receives an SE decision is considered “cleared.” The FDA will then add the cleared 510(k) to the 510(k) database, which is updated weekly. The IFU and the summary will be sent as attachments to the SE letter. The IFU will not be signed since it is considered an attachment to the SE letter. Therefore, the signature on the SE letter will apply to both the letter and the IFU.
There may be some interaction with the FDA after the AI submission to finalize any remaining items. In most cases, the FDA expects a same-day or next-day response to their inquiry to meet the 90-day target for 510(k) review.
In order to minimize the time it takes for the FDA to approve a medical device, their goal is to make a MDUFA Decision for a 510(k) within 90 FDA Days. This is the number of calendar days between the date the 510(k) was received and the date of a MDUFA decision, excluding the days the submission was on hold for an AI Request. MDUFA Decisions for 510(k) submissions include findings of substantially equivalent (SE) or not substantially equivalent (NSE).
So, how long does it take for the FDA to approve a Class II medical device? A good guidepost is six months, though some may get approval in as short as three.
Graphical explanation of the 510(k) process:
Class III devices generally follow the PMA pathway. The most involved of the FDA’s medical device approval pathways, this pathway requires scientific evidence to demonstrate safety and efficacy. While this process is more intensive than a self-registration and a 510(k), the FDA’s timeline for a Class III medical device approval is shortening, averaging eight months.
How to Reduce the Time it Takes for the FDA to Approve a Medical Device
You can’t control the FDA’s medical device approval timeline, but there are some things you can do to streamline the process:
- Prepare early: Before you do anything else, be sure you have a solid understanding of the FDA’s role in your medical device development process. Identify which FDA medical device approval pathway applies to your device so you can allocate the appropriate time, money and resources required.
- Partner with the FDA: Don’t view the FDA as a roadblock. Rather, they are a partner who is ready and willing to collaborate to get your product to market faster. While their job is to ensure safety and efficacy, they also want to get innovative products out to patients expeditiously. Form an open, collaborative relationship early—and don’t cut corners. When you establish trust, they’ll be more likely to work with you to speed up approvals.
- Write a comprehensive submission: Regardless of your device class, do your due diligence in demonstrating SE or proving safety and efficacy. Take your time writing the submission and make sure to check all the proverbial boxes.
- Get help with your prototype: Partner with a medical device prototype development expert who will ensure adherence to standards, provide proof that all risk factors were considered and guarantee a reliable and safe medical device.
You can’t predict precisely how long it will take for the FDA to approve a medical device. But by having a basic idea of the FDA’s medical device approval pathways and timelines, you can better plan, take proactive steps and ensure a smoother process. At Vantage MedTech, our medical device regulatory compliance consultants can help you navigate the complex FDA approval process. We will ensure your device meets the required regulatory controls.
Contact Vantage for more detailed information about how long it takes to get FDA approval for your medical device.
* https://www.paconsulting.com/insights/2016/gaining-competitive-advantage-in-medical-devices/


